东京医科齿科大学难治疾患研究所的鸟居晓项目副教授、清水重臣教授、顺天堂大学研究生院医学研究科的佐藤荣人专任副教授、服部信孝教授等人组成的研究团队明确了由PARK22/CHCHCHD2基因突变导致的帕金森病发病机理。相关成果已发表在EMBO Molecular Medicine在线版上。
供图:东京医科齿科大学
目前已知,帕金森病的发病与中脑黑质多巴胺能神经元的变性和脱落有关。虽然大多数帕金森病病例是散发性的,但也有因α-突触核蛋白等基因突变导致的家族性帕金森病。近年来,研究发现CHCHD2是家族性帕金森病的致病基因(PARK22),也是散发性帕金森病的风险基因,但对于帕金森病的发病机理还有许多未知的部分,改善疾患的方法也不明确。
此次研究团队明确了CHCHD2中导致多种疾患的突变——第61位苏氨酸转变为异亮氨酸的T61I突变是导致帕金森病发病的机理。
将来自小鼠的神经母细胞瘤Neuro2a细胞进行神经分化后发现,CHCHD2野生型(CHCHD2WT)会分布在线粒体中,而T61I突变型(CHCHD2T61I)虽然会一度进入线粒体,但随后又会移动到线粒体外。由于这种定位错误,CHCHD2T61I通过召集CK1ε/δ,激酶活性引起α-突触核蛋白和神经丝构成因子(NEFL)磷酸化,结果诱导形成了聚集体。
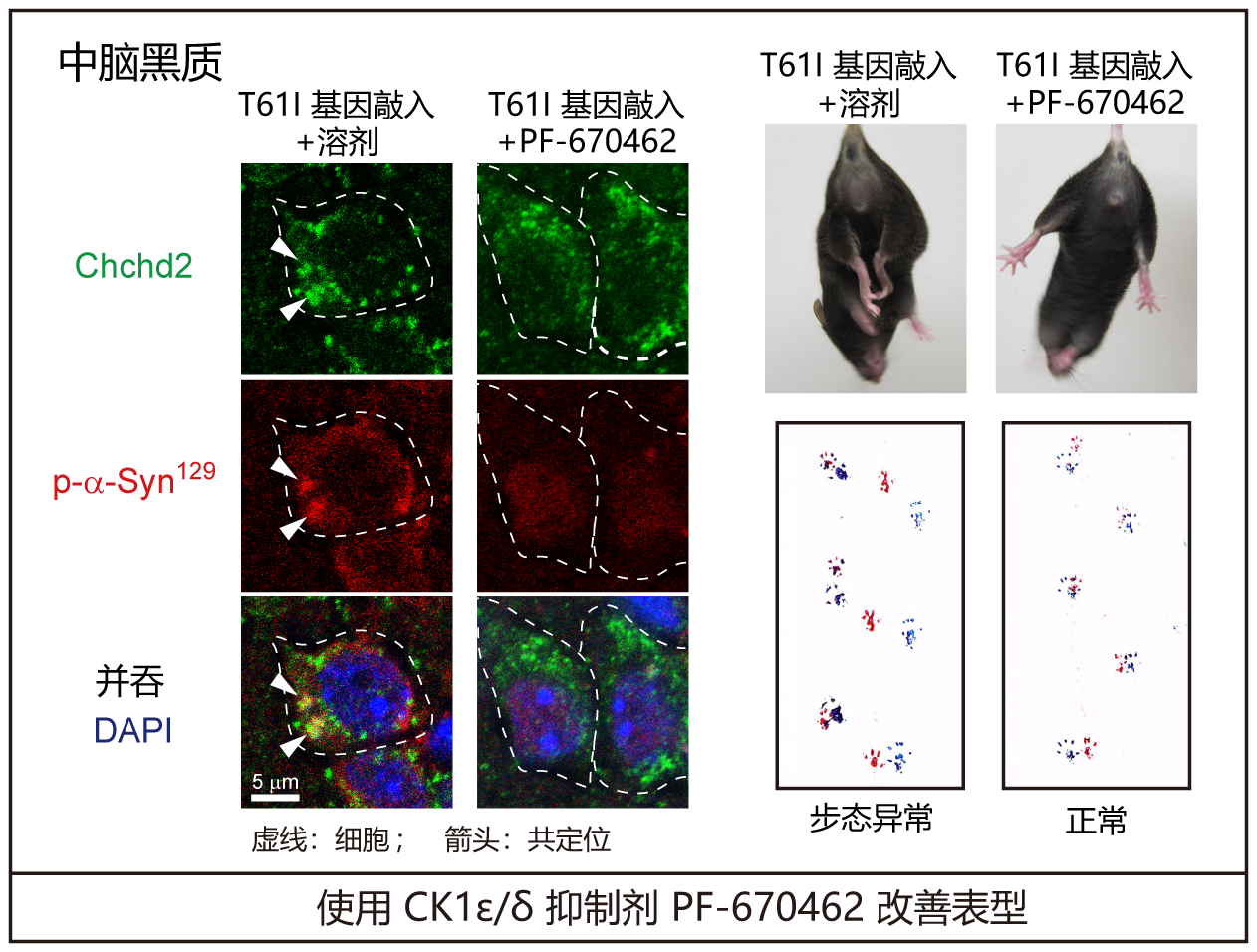
使用小鼠的分析表明,Chchd2T61I基因敲入小鼠和转基因小鼠的多巴胺能神经元中均形成了含有Chchd2T61I、CK1ε/δ、磷酸化α-突触核蛋白和磷酸化NEFL的聚集体,同时观察到神经变性特有的步态异常和运动功能障碍。在死亡的帕金森病患者的大脑和由源自患者的iPS细胞制成的多巴胺能神经元中也观察到了类似的聚集体。
为此,研究人员用CK1ε/δ抑制剂PF670462处理细胞和小鼠后,发现可显著抑制α-突触核蛋白和NEFL的磷酸化。此外,PF670462还能抑制多巴胺能神经元的细胞死亡,并改善Chchd2T61I突变基因敲入小鼠的抓握行为和步态异常等神经退行性表型。
鸟居项目副教授表示:“虽然这次我们重点就CHCHD2的T61I突变进行了实验,但我们认为这种现象还可能与目前尚不清楚病因的的散发性帕金森病有关。由于在α-突触核蛋白的磷酸化这一点上是共通的,所以我们将以此为靶点,进一步开发散发性帕金森病的治疗方法。”
原文:《科学新闻》
翻译:JST客观日本编辑部
【论文信息】
杂志:EMBO Molecular Medicine
论文:Involvement of casein kinase 1 epsilon/delta (Csnk1e/d) in the pathogenesis of familial Parkinson’s disease caused by CHCHD2
DOI:doi.org/10.15252/emmm.202317451