东京大学生产技术研究所特任研究员冈本有司(现任京都大学医学研究科特定助教)、助教梶田真司(现任福井大学学术研究院工学系部门助教)、教授合原一幸(现任东京大学特别教授)、东京大学医科学研究所教授东条有伸(现任东京医科齿科大学理事、副校长)等人组成的研究团队开发出了一种数理统计模型,可针对每个慢性髓性白血病患者,早期预测治疗药物尼洛替尼的疗效。相关研究刊发在《npj Systems Biology and Applications》上。
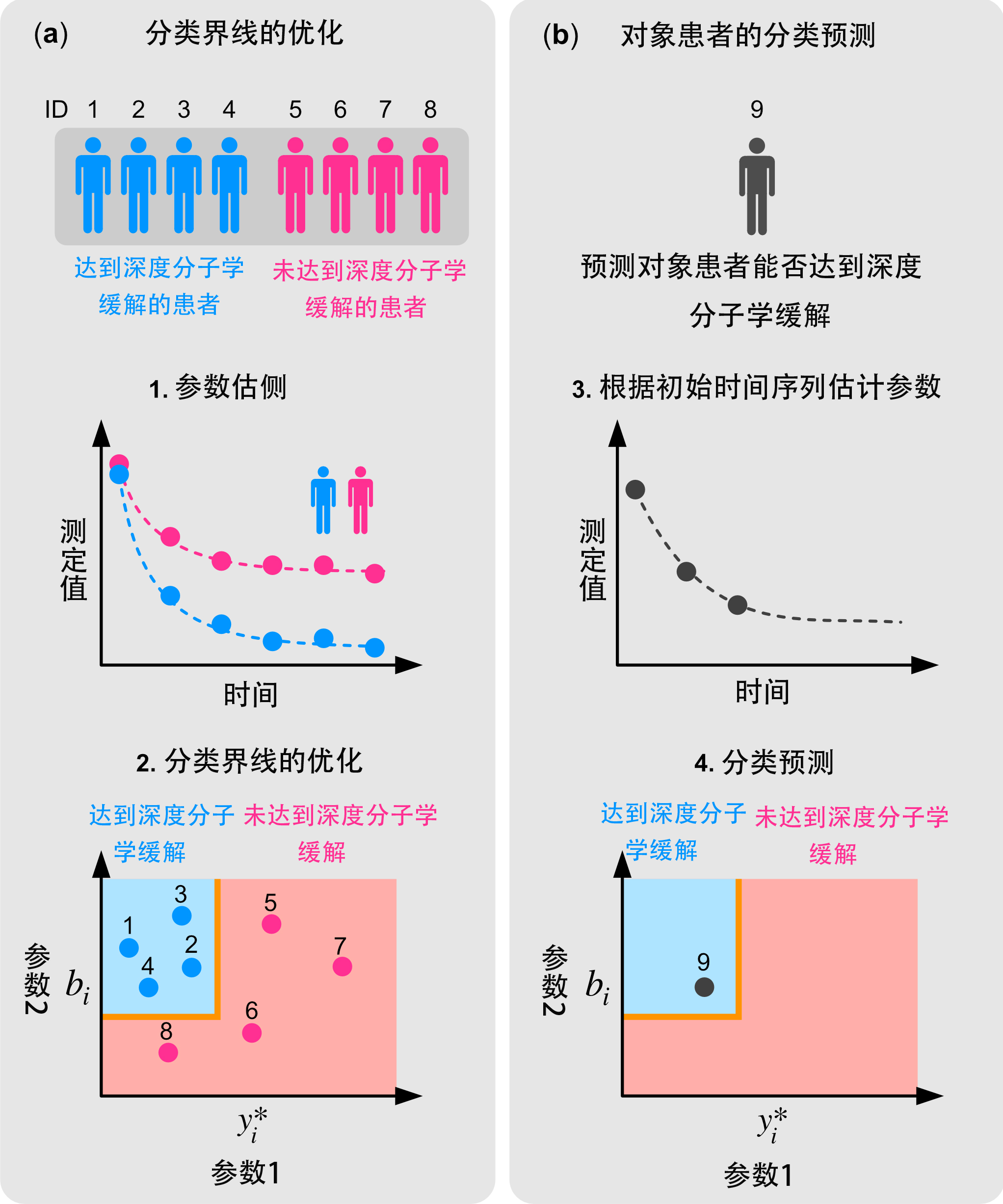
本研究提出的数理分析方法的四阶段流程图。
(a)使用CML病情相关的时间序列数据和数理统计模型,确定每个患者固有的数理统计模型参数(步骤1)。在所确定的参数空间中,通过优化明确两年内达到深度分子学缓解的患者和未达到深度分子学缓解患者的分界线(步骤2)。到此为止为学习阶段。
(b)根据从治疗开始到治疗第6个月的时间序列数据,估测对象患者的参数,以预测其在两年内是否能达到深度分子学缓解(步骤3)。在步骤2中确定的分界上绘制预测对象患者的参数,如果进入蓝色区域,则确定其两年内能达到深度分子学缓解。在上图的示例中,预测对象患者能达到深度分子学缓解(步骤4)。(供图:福井大学学术研究院工学系部门 生物应用化学讲座助教梶田真司)
慢性髓性白血病(CML)是由于白细胞、红细胞、血小板等的前期——造血干细胞的异常而导致的。由异常的造血干细胞分化而成的白血病细胞(CML细胞),具有由两个染色体之间的相互转座而形成的BCR-ABL1融合基因,该基因产生的酪氨酸激酶的异常活性导致CML细胞的无序增殖。
目前有一种名为BCR-ABL1酪氨酸激酶抑制剂(TKI)的治疗药物,可大幅减少CML细胞。虽然能改善多数患者的预后,但其效果存在个体差。
CML的治疗效果采用国际标准(IS)值来衡量。IS值是一项反映BCR-ABL1融合基因表达量的指标,可通过血液检查测定。通过治疗,如果CML细胞的量充分减少,达到深度分子学缓解,那么IS值也将充分减小。
目前,对新诊断为CML的患者使用的TKI药物包括第一代TKI伊马替尼,第二代TKI达沙替尼、尼洛替尼和博舒替尼。如果给药中的某种TKI效果欠佳,可以更换为其它TKI作为治疗选项。因此,有必要针对每个患者的TKI疗效进行早期预测。
研究团队提出了一种数理统计模型,可根据短时间序列数据,预测尼洛替尼对每个CML患者的有效性。该模型构建了一种分类方法,分别根据开始使用尼洛替尼时、使用第3个月时、使用第6个月时的IS值及外周血中的白细胞总量,来估计CML患者在使用尼洛替尼两年内能否达到深度分子学缓解状态。其中关键点是关注了外周血中CML细胞量的时间变化。
首先,使用由四个参数组成的简单常微分方程模型来表示外周血中CML细胞量和正常白细胞量的时间变化。通过改变每个患者的数据,可模拟其外周血中CML细胞和正常白细胞含量的时间变化。然而,通常的血液检查无法区分测定CML细胞和正常白细胞,为此,研究团队开发了数理统计模型,根据可测定的IS值和外周血中的白细胞总量,估计无法直接测定的外周血中CML细胞和正常白细胞的含量。
其次,使用慢性髓性白血病患者的临床试验数据,根据每个CML患者的IS值和外周血中白细胞总量的两年时间序列数据来估计出每个患者CML细胞量的常微分方程模型参数。结果表明,基于该估计参数,可将患者分类为达到深度分子学缓解的患者和未达到深度分子学缓解的患者。
最后,将患者分为学习组和疗效预测组,构建分类模型。根据学习组患者的两年时间序列数据估计出每个患者的参数值,结合其是否达到深度分子学缓解,优化并确定分类的界限。随后,根据预测组患者用药6个月的数据,估计预测对象患者的参数值,并基于分界预测其能否达到深度分子学缓解。结果显示,根据被称为MR4.5的深度分子学缓解达成标准,在使用最佳分界确定法的情况下,预测能否达到深度分子学缓解的正确率可高达94%。
此次使用的临床试验数据仅来自32名患者,且仅验证了尼洛替尼的有效性,因此今后仍需使用大规模临床试验数据,验证多种TKI的有效性。然而,通过血液检查可以预测每个患者TKI疗效的这一成果则具有重要意义。
原文:《科学新闻》
翻译:JST客观日本编辑部
【论文信息】
杂志:npj Systems Biology and Applications
论文:Early Dynamics of Chronic Myeloid Leukemia on Nilotinib Predicts Deep Molecular Response
DOI:10.1038/s41540-022-00248-3