本文根据日本东北大学成果发布资料编译整理而成

日本东北大学加龄医学研究所基因表达控制领域的冈崎庆斗助教、关根弘树讲师和本桥Hozumi教授组成的研究团队,与该研究所呼吸外科学领域的冈田克典教授、东北大学医学系研究科的铃木贵教授,以及该校信息科学研究科和Medical Megabank Organization的木下贤吾教授等人合作,共同在NRF2被激活的癌症(以下称“NRF2激活癌”)中发现了维持癌症干细胞干性所需的基因组区域。研究发现,该基因组区域通过在NRF2激活癌中发挥特异性功能并增加NOTCH3蛋白来维持癌症干细胞干性。这项研究成果有望用来有效治疗对抗癌药产生耐药性的NRF2激活癌。
背景
转录因子NRF2是通过与名为抗氧化响应序列的碱基序列结合来激活转录的蛋白质。在正常状态下,NRF2通过激活参与生物防御的各种基因来进行解毒代谢和响应氧化应激,对维持我们的健康起着重要作用(图1左)。另一方面,已知在肺癌和头颈癌中,NRF2会被特定的基因突变异常激活,导致病情恶化(图1右)。
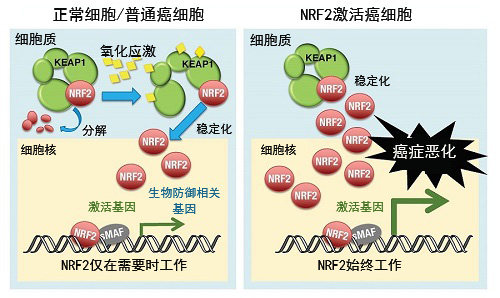
图1:在NRF2激活癌中,NRF2始终发挥作用,导致癌细胞恶化
在正常细胞和普通癌细胞中,转录因子NRF2的功能受细胞质蛋白KEAP1调控(左图)。平时KEAP1会诱导NRF2分解,抑制NRF2的功能。细胞受到氧化应激后,KEAP1便不再发挥作用,NRF2变得稳定,并发挥转录因子的功能,一举激活参与生物防御的基因簇。而在部分癌细胞中,KEAP1对NRF2的分解出现问题,NRF2始终保持稳定。这种NRF2激活癌细胞的致瘤性很强,对抗癌治疗的耐性也比较强,属于难治性癌症。
此前的研究表明,抑制被异常激活的NRF2的功能可以抑制这类癌细胞增殖,改善抗癌药的效果。不过,服用NRF2抑制剂的话,全身的正常细胞的NRF2也会被抑制,考虑到生物防御因子NRF2的重要性,这样可能会发生各种副作用。因此,需要开发能把对正常细胞的影响降到最低,同时可以有效消灭NRF2激活癌的治疗靶标。
此次的研究
研究团队在NRF2被特定基因突变异常激活的NRF2激活肺癌细胞和普通肺癌细胞中比较了NRF2的工作方式。结合名为RNA序列和ChIP序列的综合分析方法比较NRF2控制的基因表达和增强子的形成发现,NRF2的工作方式存在差异。也就是说,此次发现,在NRF2激活肺癌细胞中,NRF2通过与另一个转录因子CEBPB协同工作,来与异常的基因座结合形成增强子,并激活该基因的转录(图2)。尤其是发现,NRF2与NOTCH3基因座结合的基因组区域会作为增强子增加NOTCH3蛋白,从而增强癌症干细胞的干性。这意味着,抑制NOTCH3的话,可以抑制恶性程度较高的NRF2激活肺癌的癌症干细胞干性。
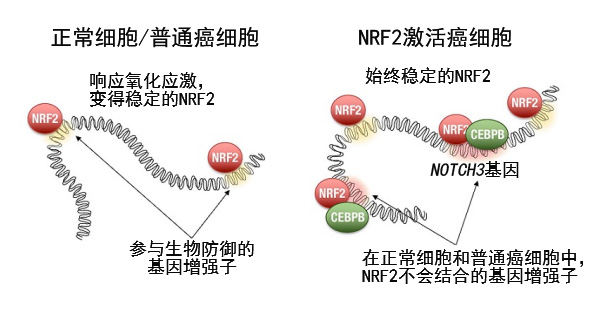
图2:在NRF2激活癌中,NRF2通过与平时不会结合的位点结合来形成增强子
调查基因组上的NRF2结合位点发现,NRF2在NRF2激活癌细胞中的结合位点与正常细胞和普通癌细胞不同。NRF2通过与另一个转录因子CEBPB协同工作,可以在NRF2激活癌中作用于特征性基因座。其中之一是NOTCH3基因座,会随着与NRF2结合形成增强子,强力激活NOTCH3的基因表达。NOTCH3能增强NRF2激活癌细胞的干细胞干性,加剧癌症恶化。
研究的意义
在NRF2激活癌中,代谢细胞膜上的药物的转运蛋白表达增强,抗癌药会快速代谢掉,这被认为是此类癌症难以治疗的原因之一。NOTCH3为膜蛋白,可以从细胞外抑制其功能,因此靶向NOTCH3的治疗战略被认为可以避免NRF2激活肺癌中增强的药物代谢转运蛋白的影响(图3)。研究团队已经通过动物实验发现,与NOTCH3胞外域发生反应的抗体对NRF2激活癌有抗肿瘤效果、在通过抑制NOTCH3来抑制癌症干细胞干性的同时使用细胞毒性抗癌药可以发挥协同作用。
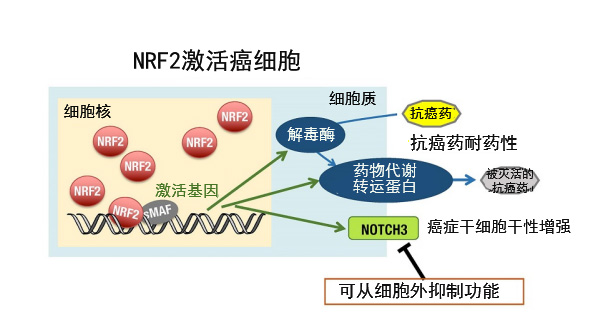
图3:对于能强力解毒代谢抗癌药物的NRF2激活癌,可以从细胞外抑制功能的NOTCH3是有效的治疗靶点
NRF2激活癌细胞中高度表达药物的解毒酶和向细胞外代谢药物的转运蛋白,对抗癌药的耐药性增强。NOTCH3为膜蛋白,可以从细胞外抑制其功能。因此,可以说NOTCH3是治疗NRF2激活癌的有效靶点。
此次研究通过明确NRF2激活癌中的特征性NRF2的功能,确定了维持癌症干细胞干性所需的基因组区域。另外还发现,在该区域的作用下产生的NOTCH3蛋白可作为NRF2激活癌这种难治性癌症的新的有效治疗靶点。NRF2激活癌可通过分析癌症的基因突变诊断,因此,此次的研究成果有望成为根据癌症的基因诊断为患者量身定制治疗方案的先驱研究成果。
论文信息
题目:Enhancer Remodeling Promotes Tumor-Initiating Activity in NRF2-Activated Nonsmall Cell Lung Cancers
期刊:Nature Communications
DOI:10.1038/s41467-020-19593-0
日语发布
编译:JST客观日本编辑部