本文根据理化学研究所成果发布资料编译而成

日本理化学研究所(以下简称“理研”)生命机能科学研究中心计算分子设计研究团队的小松辉久研究员、冲本宪明高级研究员及泰地真弘人组长等人组成的研究团队,对新冠病毒感染症(COVID-19)的致病病毒“SARS-CoV-2”的主蛋白酶(Mpro)蛋白与7种人类免疫缺陷病毒(HIV)蛋白酶抑制剂结合的过程进行了分子动力学模拟(视频)。
病毒通过使感染的细胞产生病毒蛋白来增殖。SARS-CoV-2的Mpro主要发挥“剪刀”(蛋白酶)的作用,负责在适当的位置剪断细胞产生的蛋白质并使其变得完整。Mpro与HIV的蛋白酶类似,因此有望利用现有的HIV蛋白酶抑制剂治疗SARS-CoV-2。
研究团队对7种HIV蛋白酶抑制剂分别与Mpro表面接触的过程进行了分子动力学模拟,调查了容易结合的位点分类,以及与蛋白酶活性位点结合的难易度。另外,通过解析位点的结构变化,发现这种结构的变化程度非常大,即使是在与抑制剂结合的状态下,也能呈现多种形状和排列。此次的研究成果验证了抑制剂与靶蛋白的动态结合过程,为发现候选药物分子提供了新的可能性。
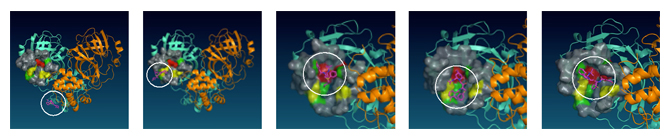
HIV抑制剂(奈非那韦=白色圆圈中的分子)与Mpro结合的过程
研究方法与成果
为推进抑制新冠病毒主蛋白酶(Mpro)的药物分子开发,研究团队通过分子动力学计算,模拟了已作为HIV抑制剂使用的7种药物分子与Mpro结合的过程。分子动力学计算利用了理研信息基础中心的HOKUSAI系统和专用计算机MDGRAPE-4A执行。
SARS-CoV-2 Mpro的立体结构根据Liu等人的报告注1)设定,作为溶剂的水分子数量接近3万个,系统的总原子数量接近10万个。将7种药物分子分别添加到含Mpro的水溶液中,并逐一对添加了不同药物分子的系统实施0.2微秒(1微秒为100万分之1秒)×28次的模拟,观察药物分子在Mpro表面的附着情况。然后,根据通过全部试验(7×28次)数据获得的药物分子与Mpro的接触数据,对Mpro表面药物容易粘附的位置(结合位点)进行了分类,并确定其中一个位置位于与Mpro的剪刀功能直接相关的位点(蛋白酶活性位点,图1)。另外,通过调查0.2微秒的结合过程,还确认了药物分子与活性位点实际结合的情形(图2)。
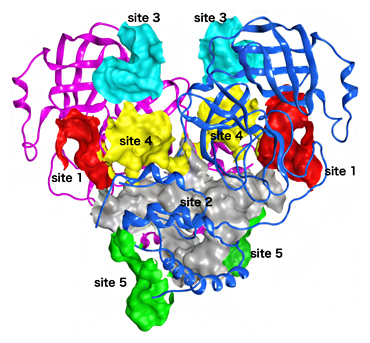
图1:SARS-CoV-2主蛋白酶(Mpro)的结合位点
Mpro作为由两个蛋白质亚基组成的同型二聚体发挥作用。根据7种药物分子的结合数据,对Mpro表面的结合位点进行了分类,显示了5个主要位点(site1-site5)。用红色显示的site1与蛋白酶活性位点重合。
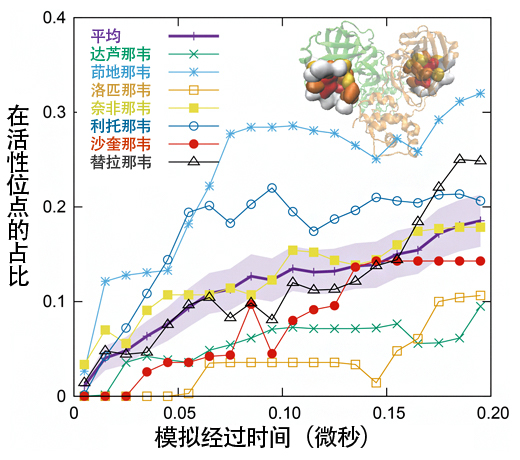
图2:药物分子与活性位点结合的过程
本图显示了在利用7种药物分子的MD模拟中,各药物分子在蛋白酶活性位点的占比随时间发生的变化。可以看出,在0.2微秒的时间里,与活性位点的结合逐渐增加。受样品数量限制,很难准确预测每种药物分子的优劣,但茚地那韦(淡蓝色)的亲和性明显高于达芦那韦(绿色)。
为捕捉与药物分子结合的Mpro的结构变化,针对在上述模拟中各药物分子最终与活性位点结合的23次试验延长了分子动力学计算,观察了1微秒内的动态。由此发现,结合位点的形状会大幅变化。另外,将3次试验的计算时间延长至6微秒后,偶尔会观察到药物分子在结合位点中改变方向(图3)。这表明,药物分子与靶蛋白的结合比以前认为的要灵活得多。
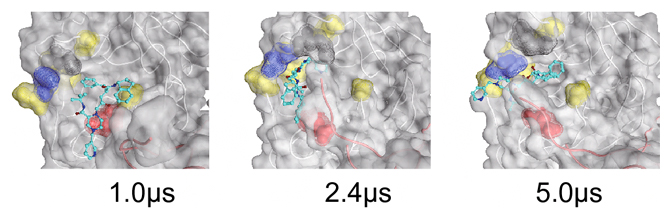
图3:在结合位点中改变方向的药物分子
对茚地那韦(用淡蓝色主链显示的棍状模型)与结合位点site-1(活性位点)的结合进行6微秒MD模拟的结果。茚地那韦的方向在1.0微秒(μs)、2.4微秒和5.0微秒时大幅改变。Mpro用空间填充模型表示,与跟茚地那韦结合有关的氨基酸残基分别用红色(第166位谷氨酸)、蓝色(第189位谷氨酰胺)、黄色(第44位半胱氨酸、第143位甘氨酸、第187位天冬氨酸、第188位精氨酸、第190位苏氨酸)和灰色(第49位甲硫氨酸)显示。可以看出,这些氨基酸残基的位置关系也在发生变化。
未来展望
今后,为获得用于探索候选药物分子和开发新药物分子的数据,研究团队预定利用更广泛的药物分子,对在SARS-CoV-2 Mpro和SARS-CoV-2中编码的其他潜在新药靶蛋白进行模拟。
注1)Title: The crystal structure of COVID-19 main protease in complex with an inhibitor N3, Entry authors: Liu, X., Zhang, B., Jin, Z., Yang, H., Rao, Z., Initial deposition on: 26 January 2020
论文信息
题目:Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation
期刊:Scientific reports
DOI:10.1038/s41598-020-74099-5
成果发布资料
编译:JST客观日本编辑部