正电子发射型计算机断层显像(Positron Emission Computed Tomography,简称PET)是核医学领域比较先进的显像手段。其在肿瘤和中枢疾病诊断方面发挥着极其重要的作用。由于PET成像可以提供三维的,全身的,动态的图像,与组织活检相比,PET显像结果更能真实的反馈肿瘤微环境的整体情况。因此,PET已经被广泛用于临床病人的诊断和筛选。但是,目前应用于肿瘤显像的正电子药物相当有限,开发新型的正电子药物,是该领域重要的任务之一。近日,日本国立研究法人量子科学技术研发机构国立放射线医学综合研究所先进核医学部张明荣教授课题组,在开发新型的多肽正电子药物方法上有了新的突破,其课题组胡宽博士研究员等人基于蛋白蛋白相互作用靶点,提出了一种“基于界面肽的PET药物”开发理论,并首先在程序性死亡受体(PD-1)/程序性死亡受体配体1(PD-L1)靶点对中验证了该方法的可行性(如图1)。
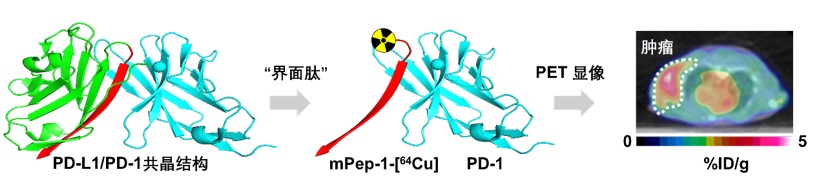
图一:利用“界面肽”策略设计靶向程序性死亡受体1(PD-1)的多肽正电子显像药物。第一步:对PD-1/PD-L1的共晶结构进行分析,找到合适的界面肽序列。第二步:对界面肽进行核素标记,稳定性研究和体外研究。第三步:在模型老鼠中研究“界面肽”的肿瘤归巢能力以及药动代力学数据。
研究背景
众所周知,多肽药物由于其良好的靶点特异性,优良的体内药代动力学性能,被认为是肿瘤核医学显像领域最有前景的一类分子。目前,已有多种多肽(类肽)正电子药物广泛应用于临床研究,如68Ga-DOTATATE, 68Ga-PSMA-617等。尽管在过去的近二十年,多肽正电子药物取得了巨大的进展,但是,全新的多肽正电子药物鲜有出现。究其原因,主要来自于缺乏寻找先导多肽的有力的方法。尽管基于高通量筛选的方法,如噬菌体展示,RNA展示,芯片技术等方法,可以从庞大的多肽库里面找到高特异性的结合靶点蛋白的多肽(2018年英国科学家Gregory P. Winter爵士因多肽和抗体的噬菌体展示技术而分享了2018年的诺贝尔化学奖)。该方法虽然十分有效,但是,耗费的时间和周期较长。更为重要的是,对于某些靶点,并不适用于高通量筛选,这就要求有其他有效的方法快速寻找多肽先导分子。
过去的数十年,科学家主要将注意力集中在癌细胞表面的受体和抗原等,因此,在开发针对这些靶点的多肽核素药物时,人们最先想到这些分子的内源性配体(图二A)。通过对配体进行改造和标记,可以得到一些效果优异的显像分子。但是,对于许多的生物标志物,尤其是细胞内的蛋白,他们往往缺乏内源性的配体,取而代之的是与之相互作用的蛋白质,这些相互作用构成了一个庞大的蛋白互作网络。目前已知人体内有超过30万对蛋白相互作用,他们参与调控细胞的生老病死。当特定的蛋白互作偏离正常轨道,就会引发疾病。因此,通过外源分子调控异常的蛋白质互作,成为了癌症治疗领域重要的方向之一。同时,这些异常的蛋白互作,也有潜力成为新的生物标志物。
蛋白蛋白相互作用(protein-protein interactions, PPIs)是由一段或者几段多肽介导的,这些多肽被称之为“界面肽”。人工合成界面肽可以一定程度上保留对互作蛋白的亲和力和特异性,因此,界面肽被已经广泛应用于PPI抑制剂的开发。但是,界面肽是否能够用于正电子显像领域,还未有相关研究进行探索(图二B)。
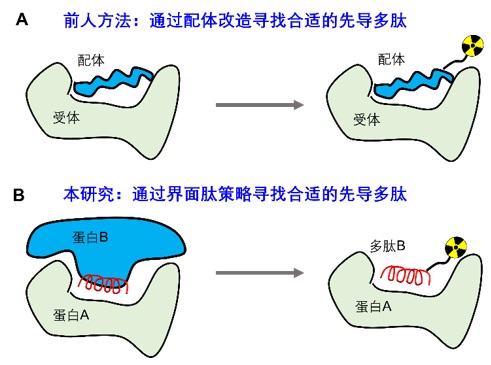
图二:两种不同的用于正电子显像的先导多肽开发策略。(A)癌细胞表面某些受体会特异性高表达,其内生的配体或类似物可以用于受体靶向的正电子显像。如血管内皮生长因子(VEGF)靶向血管内皮生长因子受体(VEGFR);(B)蛋白蛋白相互作用通常由一段或者几段关键多肽介导。通过对这些序列进行分析,可以得到一系列界面肽,这些多肽可以作为靶点识别肽的先导化合物。
研究内容
在本研究中,作者选取了PD-L1/PD-1这一对经典的蛋白相互作用,作为概念研究对象。通过对PD-1/PD-L1的共晶结构进行分析,作者发现人的PD-L1/PD-1 和 老鼠的PD-L1/PD-1的晶体结构具有极大的相似性,其中界面肽部分,PD-L1与PD-1结合的关键序列为120-131多肽,在老鼠和人中,只有V128L这一个氨基酸不同。因此,作者选取了这一段序列作为先导多肽,同时保留了这一个氨基酸的差异,合成了两条多肽,hPep-1和mPep-1。如图三所示。
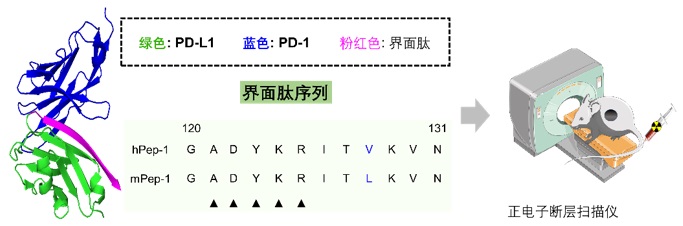
图三:通过分析PD-L1与PD-1共晶结构,认为来自PD-L1的界面肽hPep-1(人源的多肽)和mPep-1(鼠源多肽)可以作为靶向PD-1的多肽先导化合物。进而利用先导多肽对老鼠肿瘤进行PET显像。
接下来作者对这两条多肽进行了核素64Cu标记,并对标记后的稳定性进行了考察。两种多肽药物在鼠血清中表现出较强的稳定性,孵育1小时,未观察到明显的多肽降解和64Cu解离。接下来,作者对这两种药物进行了体内成像研究。选取了负载鼠黑色素瘤B16F10的老鼠作为研究模型,该模型保留了老鼠的完整的免疫系统,在肿瘤微环境中,具有大量浸润性的淋巴细胞。因此,该模型是一个非常适合PD-1PET显像的模型。显像结果如图四所示。在通过微静脉注射药物之后20分钟,观察到mPep-1-[64Cu]在肿瘤中的大量摄取,但是hPep-1-[64Cu]在肿瘤中的摄取明显低一些。在注射后40分钟,同样观察到了类似的显像。通过对PET图像进行定量分析,我们发现mPep-1-[64Cu]在肿瘤和脾脏中的摄取和保留时间要明显高于hPep-1-[64Cu],这也意味着mPep-1-[64Cu]是更好的靶向PD-1的PET显像分子。接下来,作者通过改变肿瘤移植的位置,同样观察到了类似的现象。但是在免疫缺陷老鼠中,两种多肽都不能对肿瘤进行显像。
作者进一步通过体外分布实验,验证了上述结果,并明确mPep-1-[64Cu]在肿瘤中的摄取为hPep-1-[64Cu]的1.5-2倍。为了证明mPep-1-[64Cu]为特异性结合PD-1,作者还进行了体外放射线自显影实验。通过上述结果,明确的表明了mPep-1-[64Cu]是靶向PD-1的有效的多肽显像分子。
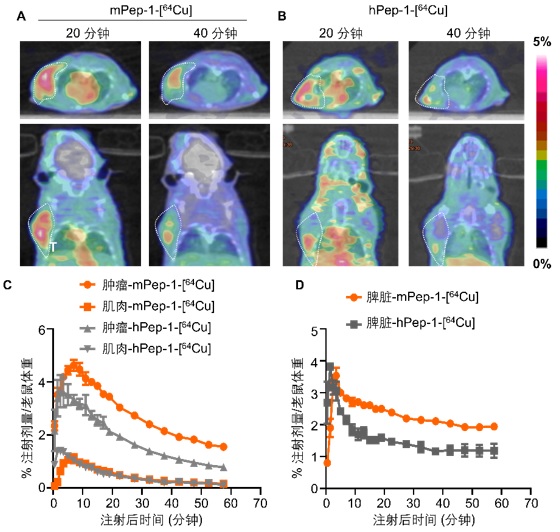
图四:(A) mPep-1-[64Cu]以及hPep-1-[64Cu]在鼠黑色素瘤B16F10中的显像结果。尾静脉注射(A)mPep-1-[64Cu]和(B)hPep-1-[64Cu]后20分钟和40分钟,肿瘤中正电子药物的富集情况。通过PET图像,对肿瘤和肌肉(C)和脾脏(D)进行时间-放射性活度关系动态统计分析。
由于hPep-1和mPep-1只有一个氨基酸的差别,但是他们的显像性能差异明显。这引发了作者的兴趣。为了探讨hPep-1和mPep-1发生差异的原因,作者测试了两种多肽对B16F10细胞的结合能力。结构表明,mPep-1具有更高的细胞结合能力,其EC50 (53.88±3.37 nM)约为hPep-1-DOTA (EC50 = 170.40±3.39 nM)的三分之一。为了厘清多肽结合能力差异的原因,作者对这两种多肽的结构进行了模拟,结果表明,两种多肽的N端表现出类似的螺旋构型,然而,C端的结构明显不同,hPep-1具有类似beta折叠的结构,然而,mPep-1的N端更像是无规卷曲(图五)。
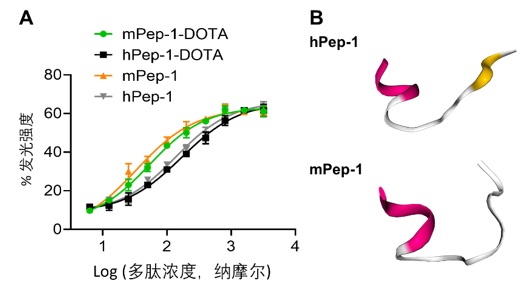
图五:(A)荧光素酶法测试多肽与PD-1结合能力。(B)通过在线工具PEP-FOLD3.5对多肽mPep-1以及hPep-1的结构进行预测。
研究结果
通过上述实验,作者证明了界面肽可以作为靶向蛋白蛋白相互作用靶点的正电子药物的先导化合物。同时,利用PD-L1/PD-1这一模型,作者首次开发了成像PD-1的多肽PET药物。值得一提的是,通过观察hPep-1和mPep-1的差异,作者也重点强调了在利用界面肽设计先导化合物时,应该考虑种属差异。最后,作者认为PET显像作为伴随诊断的工具,为多肽药物开发提供了新的评价手段。
未来展望
由于PPIs种类繁多,而且很多都可以成为癌症检测的生物标志物和治疗的靶点。因此,未来,开发靶向PPIs的PET药物意义重大。该研究提出的界面肽策略可以极大的促进该领域的发展。未来,作者也将在不同的靶点中实践该方法的有效性和普适性。
论文信息
题目: Harnessing the PD-L1 interface peptide for positron emission tomography imaging of the PD-1 immune checkpoint
杂志 RSC Chemical Biology
DOI 10.1039/D0CB00070A
文 胡宽(国立研究开发法人 量子科学技术研究开发机构)
编辑:JST 客观日本编辑部