以日本庆应义塾大学医学部生理学教室的教授冈野荣之和准访问研究员中村真理,以及该校综合医科学研究中心的特任讲师盐泽诚司等人为中心组成的研究小组,利用家族性额颞叶变性(frontotemporal lobar degeneration, 以下简称FTLD)患者的细胞培养出了iPS细胞,通过使其分化为神经细胞,搞清楚了这种病症的部分机理。
本研究着眼于家族性额颞叶变性的致病基因之一微管结合蛋白Tau(以下简称MAPT)基因的R406W突变(以下简称Tau R406W突变)。携带该突变基因时会出现与阿尔茨海默病(痴呆症)非常相似的症状。
作为比较对象,研究小组通过基因编辑技术确立了将Tau R406W基因突变修正为正常型的细胞系,以及双侧基因均携带Tau R406W突变的细胞系。另外还利用各iPS细胞制作了与大脑类似的组织(神经细胞)脑“类器官”,通过进行比较,验证了Tau R406W突变造成的神经细胞异常。
实验发现,在携带Tau R406W突变的iPS细胞源神经细胞中,Tau蛋白出现磷酸化,且局部存在异常,另外还观察到了神经轴突变性等。此外确认,这些表型可以通过微管稳定剂抑制。众所周知,Tau蛋白与阿尔茨海默病等多种神经退行性疾病有关,此次发现的机制有望成为新的病理模型,用来开发能有效抑制这些异常的微管稳定剂等新型治疗药物。
FTLD是老年期随着大脑额叶和颞叶的神经细胞死亡而引起的痴呆症之一。阿尔茨海默病主要表现为记忆障碍,而FTLD的特征是会引起性格变化和行为异常等。无论哪种痴呆症,在病理学上都出现了Tau蛋白的异常积累,被认为与疾病发病有关,不过一直不清楚详细的机制。
在家族性FTLD中,编码该Tau蛋白的MAPT基因有时会发生基因突变,此前已报告过各种突变,已知携带名为Tau R406W的氨基酸突变时,会出现与阿尔茨海默病非常相似的症状。
本研究培养了仅MAPT基因单侧携带Tau R406W突变的患者的iPS细胞(杂合突变型)。作为比较对象,还培养了不携带突变的人源iPS细胞(健康人)、利用基因编辑技术修正了杂合突变型患者的突变基因的iPS细胞(突变修正型),以及双侧基因均携带Tau R406W突变的iPS细胞(纯合突变型)。研究小组利用这些iPS细胞分别制作了与大脑相似的组织——脑“类器官”,并详细分析了其神经细胞。
分析发现,在Tau R406W突变神经细胞中,①Tau蛋白变成异常的低磷酸化状态;②与不携带Tau R406W突变的对照组相比,Tau蛋白片段增加;③Tau蛋白的片段是被蛋白质裂解酶——钙蛋白酶裂解的。
另外,正常情况下Tau蛋白存在于神经细胞的轴突部分,但在携带该突变的患者的iPS细胞源神经细胞中,细胞体及树状突起中存在的Tau蛋白的比例增加。此外还确认,在携带Tau R406W突变的神经细胞中,轴突被切碎(图1),细胞器之一的线粒体的运输发生异常。线粒体运输异常的情况在添加微管稳定剂后恢复正常,因此这种情况被认为是由微管的不稳定造成的。
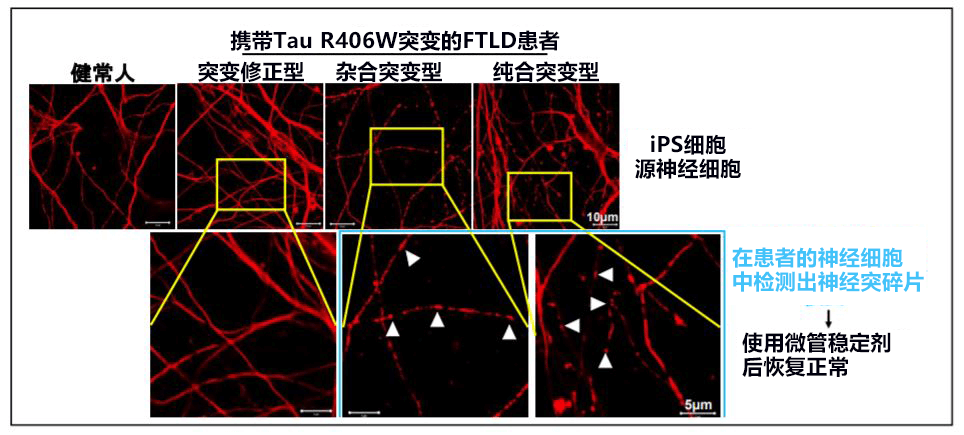
【图1】红色染色部分为轴突
根据此次的研究结果可以认为,神经细胞的Tau蛋白异常和线粒体运输异常是携带Tau R406W突变的FTLD的部分病症。
本研究明确了编码Tau蛋白的MAPT基因中发生Tau R406W突变的神经细胞出现的异常(图2)。已知Tau蛋白异常与阿尔茨海默病等多种神经退行性疾病有关。此次的研究成果有望用来开发治疗Tau蛋白引起的疾病的药物和抑制疾病发展的药物。
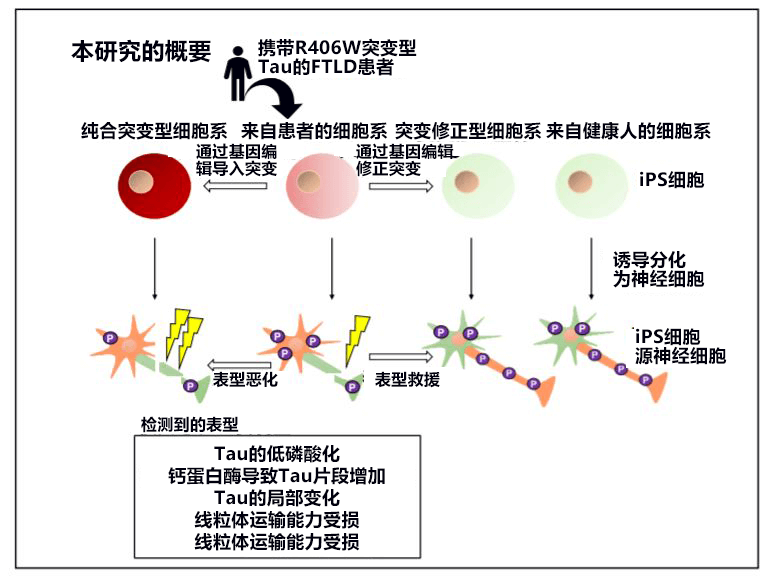
【图2】编码Tau蛋白的MAPT基因中携带Tau R406W突变的神经细胞发生的异常
相关研究成果已于2019年9月19日发布在国际干细胞研究学会(ISSCR)的官方期刊《Stem Cell Reports》的网络版上。
论文题目:Pathological progression induced by the frontotemporal dementia-associated R406W tau mutation in patient-derived iPSCs
文:JST客观日本编辑部翻译整理